Revealing an unexpected chemical reactivity of boron nitride to H2O and alkane molecules
Abstract
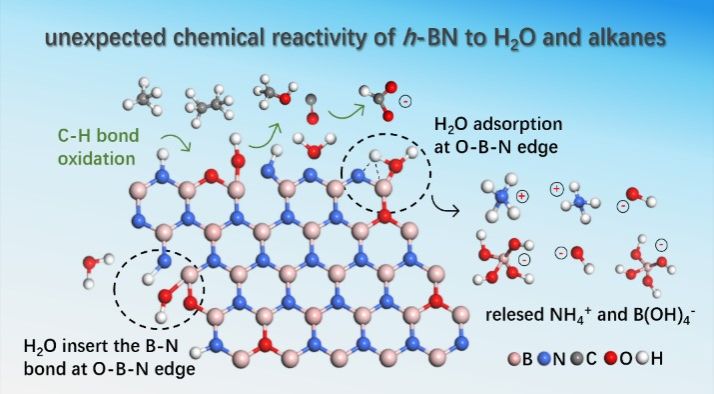
Herein, substantial experimental evidence reveals an unexpected chemical reactivity of inert h-BN to H₂O, CH₄, and C₂H₆ under mild conditions. H₂O molecules dissociate at B-N edge sites, and insert into B-N bond even under ambient conditions. Detailed spectroscopic characterization shows this process protonates nitrogen sites and hydroxylates boron sites, forming N-H and B-OH groups. Ultimately, the NH₄⁺ and B(OH)4- ions are released into water as the final products of nitrogen protonation and boron hydroxylation. This reactivity is significantly enhanced at oxygen-doped B-N edges. Theoretical simulations reveal that strong orbital interactions between the H (1s) orbitals of H₂O and the B (2p)/N (2p) orbitals of B-N edge produce significant chemical stress at the adsorption sites, promoting the dissociation and subsequent insertion of H₂O into the B-N bonds. Furthermore, we show that CH₄ and C₂H₆ can be oxidized to CO and trace CH3OH in water over boron nitride at mild temperatures without additional oxidant. The ¹⁸O isotope-tracing experiment confirms the oxygen in the boron nitride matrix is responsible for the activation and oxidation of the C-H bond of CH4 and C2H6. Simultaneously, the released NH₄⁺ and B(OH)4- ions provide a reaction microenvironment enabling thermodynamically spontaneous hydration of formed CO to formate. These findings fundamentally challenge the long-standing paradigm of h-BN as a chemically inert material under mild conditions.


